Comment les compléments alimentaires sont-ils encadrés ?
Vous vous demandez comment les compléments alimentaires sont encadrés ? Est-ce que leur sécurité, leur qualité et leur efficacité est contrôlée ? Qui peut vendre des compléments alimentaires ? Mise sur le marché, allégations, étiquetage, nutrivigilance … Découvrez ici tout ce qu’il y a à savoir sur la règlementation des compléments alimentaires.
- Un cadre complet
- Leur mise sur le marché
- La revendication de leur efficacité
- Leur étiquetage
- Les exigences qualité
- La surveillance post-commercialisation
- La règlementation bio
- Leur composition
Que couvre la règlementation des compléments alimentaires ?
Les compléments alimentaires sont strictement encadrés au niveau français et européen.
En tant que denrées alimentaires, ils doivent respecter l’ensemble de la règlementation alimentaire française et européenne.
Un cadre spécifique supplémentaire s’applique également aux compléments alimentaires.
La règlementation française et européenne des compléments alimentaires encadre ainsi :
- Leur composition ;
- Les allégations réalisées sur l’efficacité des ingrédients ;
- Les modalités de leur mise sur le marché ;
- Leur surveillance post-commercialisation.

Plus de 20 ans de règlementation pour une sécurité optimale
Fin des années 80
Apparition des 1ers compléments alimentaires en France
1996
Décret 96-307 du 10/04/96 :
1ère définition officielle du terme « Complément alimentaire »
2002
Directive 2002/46/CE relative aux compléments alimentaires.
Directive fondatrice de la règlementation européenne des compléments alimentaires
2006
Décret 2006/352 du 20/03/06
Déclaration obligatoire des compléments alimentaires avant la mise sur le marché français
Arrêté du 9 mai 2006
Définition de doses maximales françaises en vitamines et minéraux
Règlement (CE) 1924/2006
Relatif aux allégations nutritionnelles et de santé portant sur les denrées alimentaires
2006
Règlement (CE) 1925/2006 1ères procédures de restriction ou d’interdiction européenne pour les plantes et substances
2010
Mise en place de la procédure de Nutrivigilance en France en application de la loi Hôpital, patients et territoires (article 109)
2012
Règlement (UE) 432/2012 Etablissant une liste des allégations autorisées pour les vitamines et minéraux
2014
Arrêté Plantes Etablissant une liste de 540 plantes autorisées dans les compléments alimentaires
En fonction des plantes, il établit des doses maximales, des précautions d’emploi et des substances à surveiller.
Liste BelFrIt (Belgique France Italie)
Liste sans valeur juridique de plus de 1 000 plantes utilisées dans les compléments alimentaires
2016
Mise en place de la télédéclaration
La directive 2002/46/CE : Une définition harmonisée des compléments alimentaires
« On entend par «compléments alimentaires», les denrées alimentaires dont le but est de compléter le régime alimentaire normal et qui constituent une source concentrée de nutriments ou d’autres substances ayant un effet nutritionnel ou physiologique seuls ou combinés, commercialisés sous forme de doses, à savoir les formes de présentation telles que les gélules, les pastilles, les comprimés, les pilules et autres formes similaires, ainsi que les sachets de poudre, les ampoules de liquide, les flacons munis d’un compte-gouttes et les autres formes analogues de préparations liquides ou en poudre destinées à être prises en unités mesurées de faible quantité »
Article 2 de la directive 2002/46/CE
La mise sur le marché des compléments alimentaires
Un complément alimentaire est un produit très encadré par la règlementation française et européenne. La directive 2002/46/CE portant sur les compléments alimentaires est transcrite en droit français avec le décret 2006-352. Ce décret français précise les modalités de mise sur le marché d’un complément alimentaire en France.
Une notification préalable obligatoire
Pour être commercialisé en France, un complément alimentaire doit préalablement avoir fait l’objet d’une déclaration de mise sur le marché auprès de la Direction Générale de l’alimentation (DGAL). Cette déclaration, obligatoire depuis mars 2006, permet de faciliter la surveillance du marché et le contrôle des produits.
Chaque déclaration de mise sur le marché d’un produit est examinée par la DGAL sous l’angle de la conformité du produit, de la sécurité des consommateurs et du respect des exigences réglementaires. Tout fabricant ou importateur, responsable de la mise sur le marché français d’un complément alimentaire, doit donc déclarer sa mise sur le marché auprès de la DGAL avant de pouvoir commercialiser son produit.
Depuis le 26 avril 2016, la déclaration des compléments alimentaires se fait en ligne par le biais de la téléprocédure. L’outil actuel de télédéclaration s’appelle Compl’Alim.
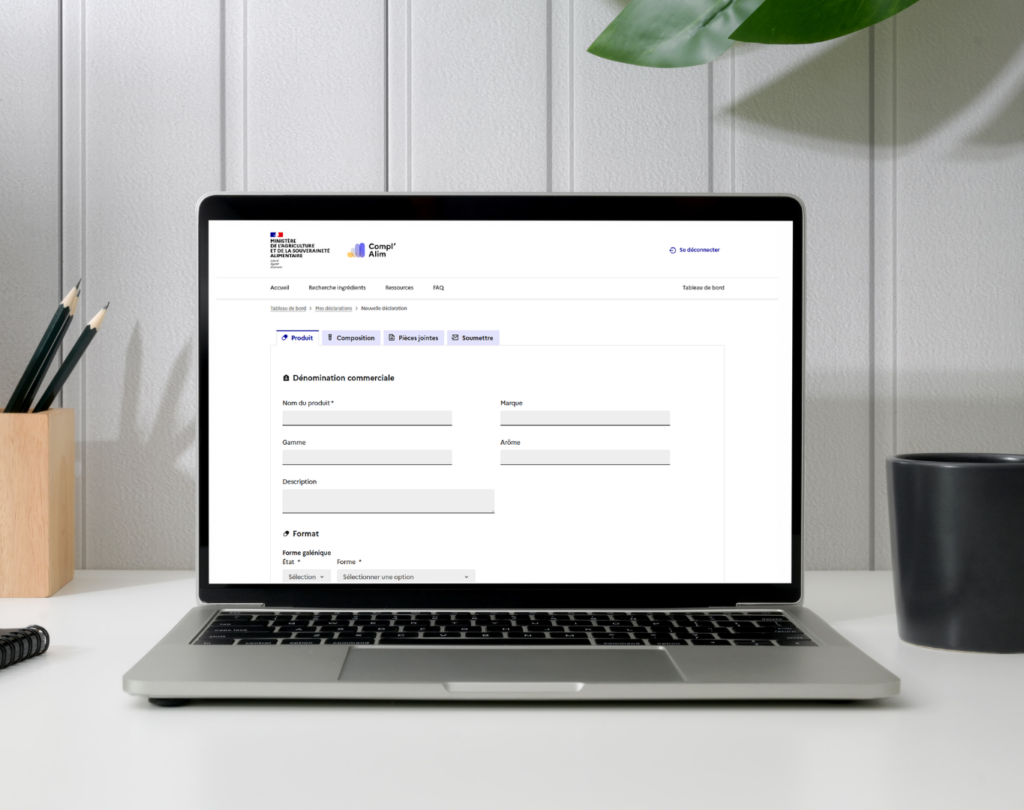
Les principales informations transmises au moment de la télédéclaration




L’objectif de la DGAL est de valider, grâce aux informations saisies, la conformité réglementaire du produit et sa sécurité.
Dès transmission de la déclaration, la DGAL dispose d’un délai de 2 mois pour l’examiner et faire un retour sur celle-ci. Si les autorités n’ont aucune observation ou objection sur la déclaration, une attestation de déclaration est alors mise à disposition du déclarant.
Télédéclaration = Plus de transparence pour le consommateur
Depuis 2017, il est également possible de consulter en ligne la liste des produits télédéclarés. Cette nouvelle possibilité permet au consommateur de vérifier si un produit a fait l’objet d’une télédéclaration. Cette liste reprend la description du produit, les ingrédients, les mises en garde et précautions d’emploi.
Attention, les produits déclarés avant la mise en place de la télédéclaration en ligne (avril 2016), ne sont pas repris dans cette liste.
2 types de déclarations
Il existe deux types de procédures de déclaration, définies dans le décret 2006-352, qui dépendent de la composition du produit :

Procédure Article 15 (ou déclaration directe) :
Le produit est conforme à la réglementation française, les ingrédients ajoutés dans le produit sont listés dans les listes positives françaises. L’opérateur déclarant un produit en article 15 peut choisir de le mettre sur le marché dès que la procédure de déclaration est finalisée, sans nécessairement attendre le retour des autorités sur la conformité du produit.

Procédure Article 16 (ou procédure de reconnaissance mutuelle) :
Le produit ou l’ingrédient qui fait l’objet de la reconnaissance mutuelle est légalement commercialisé dans un autre Etat membre de l’Union européenne (par exemple la Belgique, l’Italie, la Croatie…). Un produit en article 16 ne peut pas être mis sur le marché sans avoir reçu le retour des autorités dans un délai maximum de 2 mois.
Compléments Alimentaires et Allégations Nutritionnelles et de Santé
Images, éléments graphiques, conseils en magasin, symboles, écritures, les allégations nutritionnelles et de santé ont plusieurs formes mais un seul objectif : mettre en avant les caractéristiques particulières d’un produit. Au cœur du marketing des compléments alimentaires, elles sont strictement encadrées par la réglementation européenne.
Qu’est-ce qu’une Allégation Nutritionnelle et de Santé ?

Les allégations nutritionnelles et de santé sont des mentions ou des images valorisant des denrées alimentaires sur le plan nutritionnel, « Riche en Vitamine C », ou de la santé, « La vitamine C contribue à réduire la fatigue ».
Elles sont définies dans le Règlement (CE) 1924/2006 comme « tout message ou toute représentation, non obligatoire, y compris une représentation sous forme d’images, d’éléments graphiques ou de symboles, qui affirme, suggère ou implique qu’une denrée alimentaire possède des caractéristiques particulières ».
Ce règlement s’applique à toutes les communications à caractère commercial sur un produit à destination du consommateur final, à l’écrit (étiquetage, support de communication) comme à l’oral (publicité, conseils en magasin…).
Les grands principes du règlement Allégations



Les Allégations Nutritionnelles
Une allégation nutritionnelle correspond à un message ou une représentation graphique qui affirme suggère ou implique qu’une denrée alimentaire contient ou ne contient pas une certaine quantité d’énergie, de nutriments ou d’autres substances ayant un effet nutritionnel. Les allégations nutritionnelles correspondent aux allégations du type « Source de Fer », « Riche en protéines », « Sans sucre ajouté » …
Le règlement (CE) 1924/2006 contient une liste fermée d’allégations nutritionnelles autorisées. Pour utiliser une allégation nutritionnelle, il faut que celle-ci soit définie en annexe du règlement et réponde à ses conditions d’utilisation spécifiques.
Pour pouvoir utiliser une allégation nutritionnelle sur un complément alimentaire, le produit doit contenir, par dose journalière, une quantité significative de l’ingrédient sur lequel porte l’allégation. Par exemple, pour alléguer « Source de Vitamine C », le complément alimentaire doit apporter 15 % des valeurs nutritionnelles de référence en Vitamine C par dose journalière. Les valeurs nutritionnelles de référence sont définies en annexe 13 du règlement (UE) 1169/2011.
Les Allégations de santé

Une allégation de santé correspond à un message ou une représentation graphique qui affirme, suggère ou implique que la consommation d’un ingrédient peut avoir des bienfaits sur la santé, par exemple « Le magnésium contribue à réduire la fatigue ».
Pour pouvoir être utilisée, une allégation de santé doit être autorisée. En 2012, une première liste de 222 allégations fonctionnelles génériques portant principalement sur des nutriments et des substances a été publiée dans le règlement (UE) 432/2012. Celles-ci peuvent être utilisées à condition de bien répondre aux conditions d’utilisations mentionnées (quantités, mentions…).
Pour pouvoir obtenir une nouvelle allégation, un opérateur doit déposer un dossier scientifique auprès de la Commission européenne. Ce dernier sera étudié par l’autorité européenne de sécurité des aliments. Si son avis est favorable, l’allégation pourra être autorisée par la Commission européenne.
Les allégations de santé portant sur les plantes ont été mises en attente d’une décision par la Commission européenne.
La Commission a établi une liste de 2 078 allégations Plantes en attente. Leur utilisation est tolérée sous réserve, pour l’opérateur, de pouvoir justifier par des données scientifiques la véracité de l’allégation.
Au cours de cette période transitoire, la Commission européenne doit définir des lignes directrices permettant l’évaluation de ces ingrédients.
En savoir plus
Retrouvez l’ensemble des allégations de santé utilisables sur le site de la DGCCRF.
L’étiquetage des compléments alimentaires
L’étiquetage d’un produit est un élément déterminant dans l’orientation du consommateur au moment de l’acte d’achat. Véritable outil de promotion, il informe également le consommateur sur la nature du produit et ses caractéristiques. De nombreuses mentions sont indispensables pour assurer cette information correcte du consommateur et sont donc exigées par la réglementation.
Les compléments alimentaires sont régis par les mêmes règles d’étiquetage que les denrées alimentaires. À celles-ci s’ajoutent quelques particularités.
Les mentions obligatoires pour toutes les denrées alimentaires
Les mentions obligatoires pour l’étiquetage des denrées alimentaires sont listées en article 9 du Règlement (UE) n°1169/2011 (Règlement INCO) :
- La dénomination légale de vente : « Complément alimentaire », qui doit complétée de la mention « à base de… », par exemple « Complément alimentaire à base de plante », ou encore « … à base de vitamines et minéraux » ;
- La liste des ingrédients, doit être précédée de la mention « Ingrédients » ;
- La mise en avant des allergènes dans la liste des ingrédients, soit par une mise en gras soit en les soulignant ;
- La quantité nette, doit se trouver dans le même champ visuel que la dénomination légale ;
- Les conditions particulières d’utilisation, telles que les précautions particulières d’emploi liées à la présence de certaines plantes ou nutriments. Par exemple, l’arrêté Plantes exige l’ajout de précautions d’emploi spécifiques ; un produit contenant du Basilic devra étiqueter « Déconseillé aux enfants. Ne pas utiliser de façon prolongée » ;
- Date de durabilité minimale ou date limite de consommation et numéro de lot ;
- Conditions de conservation et mode d’emploi : la portion journalière, le moment de la prise, par exemple « Une ampoule à prendre le matin dans un grand verre d’eau » ;
- Nom/raison sociale et adresse du responsable de la mise sur le marché.

L’étiquetage des quantités d’actifs

Les compléments alimentaires sont exemptés de déclaration nutritionnelle, ils doivent uniquement indiquer les quantités en nutriments, en plantes ou en substances présentes par portion journalière recommandée. Par exemple un complément alimentaire à base de guarana et de vitamine C, devra présenter sur son étiquetage la quantité de guarana et la quantité de vitamine C (avec le % de la valeur nutritionnelle de référence) par portion journalière. L’étiquetage de la quantité de plantes peut être faite en équivalent plante sèche (EPS) qui correspond à la quantité de plante sèche mise en œuvre pour fabriquer un extrait. Plus l’EPS est élevé plus la quantité de plante utilisée pour une portion journalière est importante.
Des mentions spécifiques aux compléments alimentaires
La directive 2002/46/CE qui encadre les compléments alimentaires apporte d’autres mentions obligatoires à apposer. Ce sont le nom des catégories de nutriments, substances ou plantes, la portion journalière recommandée, un avertissement contre le dépassement de la portion journalière, une déclaration visant à éviter que les compléments alimentaires ne soient utilisés comme substituts d’un régime alimentaire varié et l’avertissement suivant « Tenir hors de portée des jeunes enfants ».
A cela peut s’ajouter des mentions spécifiques en fonction de la composition du produit, par exemple des mentions liées à la présence de caféine, d’édulcorants ou encore de certaines plantes …
Récapitulatif des mentions d’étiquetage des compléments alimentaires

Quel encadrement de la qualité des compléments alimentaires ?
Le développement d’un complément alimentaire doit prendre en compte, dès sa conception, des exigences de qualité et de sécurité. Cette étape est fondamentale car elle conditionne la qualité et la pérennité du produit.
La charte de qualité des compléments alimentaires
L’ensemble des recommandations développées dans notre Charte de Qualité est issu de la mise en œuvre du règlement 852/2004/CE, notamment son annexe II relative aux dispositions générales d’hygiène pour tous les exploitants du secteur alimentaire.
L’Analyse HACCP

La méthode HACCP, rendue obligatoire par le règlement (CE) 852/2004 sur l’hygiène des denrées alimentaires, consiste en une analyse exhaustive des dangers, de leur gravité et de leur fréquence d’apparition associés à chaque étape de la conception des produits jusqu’à leur livraison.
De cette analyse découle l’adoption de mesures de maîtrise appropriées et préventives et la définition des mesures de maîtrise critiques à mettre en place, incluant les méthodes de surveillance de ces mesures critiques, pour garantir la sécurité et la qualité des produits. L’objectif de l’HACCP est d’amener les professionnels à maîtriser efficacement les dangers susceptibles d’affecter la santé du consommateur. L’HACCP vous aide à mettre en place une gestion et une surveillance efficaces des dangers évalués comme inacceptables en terme d’impact sur la santé du consommateur. Une étude HACCP devra être appliquée dès les premiers stades du process de développement du produit afin de minimiser les risques potentiels et même de les éliminer.
Le contrôle qualité
Le contrôle qualité doit être appliqué à la fois :
- aux articles de conditionnement : chaque article de conditionnement doit se conformer aux exigences réglementaires du règlement CE n°1935/2004 et du décret n°92-631 du 8 juillet 1992 et à ses spécifications. Le packaging final doit porter les informations nécessaires et les mentions spécifiques dans la forme et à l’endroit requis. Une personne habilitée doit s’assurer que les emballages correspondent bien à la réglementation.
- aux ingrédients : leur contrôle permet de garantir la conformité des ingrédients aux spécifications requises. Ils sont adaptés à la nature des ingrédients et à leurs fonctions et cas d’emploi. Ils sont effectués à l’arrivée sur le site de production ou au départ du site du fournisseur. Aussi, tout lot d’ingrédient ne respectant pas les critères de pureté ne sera pas inclus dans le cycle de production. Toutes les étapes de la réception des matières à la libération des produits fabriqués doivent faire l’objet d’enregistrements permettant de garantir la traçabilité.

L’Assurance Qualité
L’assurance qualité comprends des audits et revue de direction, la gestion des produits non conformes, la gestion de la documentation, l’identification produit, la traçabilité amont et aval, la formation et qualification du personnel, la métrologie…
La politique de qualité et de sécurité doit intégrer le respect des réglementations relatives à l’hygiène des denrées alimentaires. L’hygiène y est définie comme étant : « L’ensemble des mesures et les conditions nécessaires pour maîtriser les dangers et garantir le caractère propre à la consommation humaine d’une denrée alimentaire compte tenu de l’utilisation prévue. »
Exemple de processus de fabrication avec les contrôles de routine
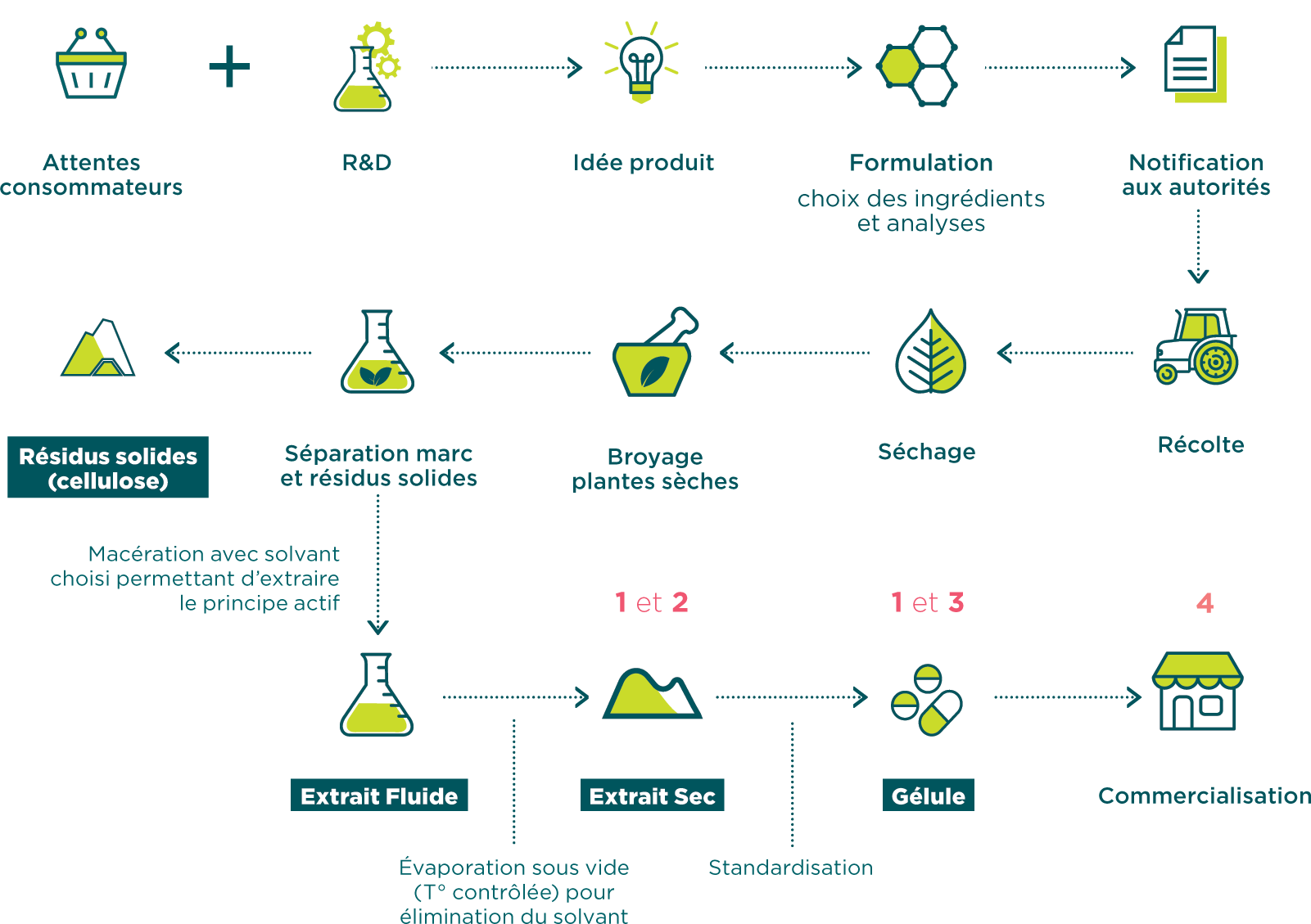
La surveillance post-commercialisation des compléments alimentaires
Le dispositif de Nutrivigilance, instauré en 2010 par le décret n°2010-688, est un système de veille sanitaire qui a pour objectif de contribuer à renforcer la sécurité du consommateur en identifiant d’éventuels effets indésirables liées à la consommation de certaines denrées alimentaires. Comment est-il mise en œuvre ? Quelles conclusions peut-on en tirer pour les compléments alimentaires ?

Quel objectif ?
Ce dispositif permet de faire remonter à l’agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail (l’Anses) les effets indésirables survenus suite à la consommation de certaines denrées alimentaires. Ce dispositif permet ainsi aux autorités de surveiller la sécurité des produits sur le marché et d’ajuster, si besoin, leur encadrement. Dans ce cadre l’Anses analyse l’imputabilité de l’effet indésirable et sa gravité.
Le dispositif de nutrivigilance concerne différentes catégories de produits dont les compléments alimentaires mais également les Novel food, les aliments enrichis et les denrées destinées à une alimentation particulière.
Qui déclare ?
- Les professionnels de santé
- Les distributeurs et fabricants
- Les particuliers
Comment déclarer ?
Les déclarations s’effectuent sur un site internet dédié en remplissant le formulaire en ligne.
Les informations à fournir sont le nom complet du produit, le sexe, l’âge et les antécédents du consommateur, les dates de prises et d’arrêt, l’effet indésirable (description, date d’apparition, évolution), et les autres étiologies possibles (médicaments, examens complémentaires, facteurs de risque…).

Quelles suites ?
Les déclarations sont ensuite analysées par l’Anses avec l’appui d’experts médicaux qui étudient les points suivants : gravité du signalement, composition du produit, regroupement avec les signalements précédents, imputabilité ou non de l’effet indésirable au produit. Les conclusions des analyses sont ensuite remises aux ministères concernés pour la mise en place de mesures de gestions adaptées. En fonction du nombre de cas, de leurs gravités et de leur imputabilité, l’Anses peut décider de s’auto-saisir afin de réaliser une évaluation plus complète et approfondie des risques liés à la consommation de certains produits ou de certains ingrédients.
Comment éviter la survenue d’effet indésirable ?
Retrouvez tous nos conseils pour la bonne consommation des compléments alimentaires
La réglementation Bio
Les produit biologiques sont encadrés par le règlement (UE) 2018/848. Ce texte définit les règles à suivre à toutes les étapes de production, transformation, étiquetage, contrôle et d’importation en provenance de pays tiers.
Un produit biologique doit ainsi répondre aux règles générales suivantes :
- Pas d’utilisation d’OGM ;
- Pas de pesticides chimiques de synthèse (insecticides, fongicides de synthèse…) ;
- Interdiction d’incorporer un même ingrédient bio et non bio dans un même produit ;
- Les préparations bio et non bio doivent être séparées dans l’espace et dans le temps ;
- Être composé majoritairement (>50%) d’ingrédients agricoles dont 95% doivent être issus de l’agriculture biologique. L’eau et le sel ne sont pas pris en compte dans le calcul du pourcentage bio. Les 5% restants sont des ingrédients agricoles inscrit dans le règlement (CE) 889/2008 (Annexe IX). Les composants non agricoles, qui sont donc <50%, doivent être inclus dans la liste des additifs autorisés en annexe VIII du règlement (CE) 889/2008. Attention, certains additifs sont suivis d’un astérisque dans le règlement, ce qui signifie qu’ils doivent être compatibilisés dans la proportion d’ingrédients d’origine agricole.

Spécificités des compléments alimentaire bio, le cumul des 2 réglementations :

- Additifs et les auxiliaires technologiques : pour être bio, un complément alimentaire ne doit contenir que les additifs qui sont autorisés à la fois dans les compléments alimentaires par la Catégorie 17 de la réglementation des additifs – Règlement (CE) 1331/2008 – et dans les produits bio conformément à l’annexe VIII du règlement (CE) 889/2008.
- Nutriments et substances : le Règlement bio précise que « les minéraux (y compris les oligo-éléments), vitamines, acides aminés et micronutriments, sont autorisés uniquement si leur emploi, dans les denrées alimentaires dans lesquelles ils sont incorporés, est exigé par la loi ». Or la réglementation des compléments alimentaires n’exige aucun ajout, il n’est donc pas possible d’ajouter des vitamines ou minéraux dans un complément alimentaire bio. La seule solution pour avoir un complément alimentaire bio apportant une source de vitamines ou de minéraux est d’apporter le nutriment par le biais d’une plante titrée en celui-ci, par exemple un complément alimentaire bio à base d’Acérola titrée en Vitamine C.
La composition des compléments alimentaires
Pour pouvoir être utilisé dans un complément alimentaire, un ingrédient actif doit être formellement autorisé.
Un encadrement national
La composition des compléments alimentaires est principalement encadrée au niveau national par chaque Etat membre de l’Union européenne.
Les Etats membres fixent ainsi la liste des ingrédients qu’ils autorisent sur leur sol, ainsi que les précautions d’emplois et doses associées. Seuls les vitamines et minéraux sont partiellement encadrés au niveau européen.
Principaux textes encadrant la composition des compléments alimentaires
- L’arrêté plantes du 24 juin 2014
- L’arrêté substances du 26 septembre 2016
- Le règlement (CE) 1170/2009 sur les vitamines et minéraux
- L’arrêté du 9 mai 2006 sur les doses de vitamines et minéraux
- Les listes de plantes, substances, algues, lichens et huiles essentielles déclarables en article 15 établies par la DGCCRF
A ces listes, s’ajoutent l’utilisation de la reconnaissance mutuelle.
Consulter notre page dédiée
Pour en savoir plus sur la manière dont la composition des compléments alimentaires est encadrée, consultez notre page dédiée.
Les nouveaux aliments : un encadrement spécifique
Avec la mondialisation des échanges et les développements technologiques, de plus en plus de nouveaux aliments – Novel Food – font leur apparition sur le marché européen. Comment sont-ils encadrés ? Synadiet vous répond.

Qu’est-ce qu’un Novel Food ?
Un Novel Food ou Nouvel aliment est un aliment ou un ingrédient alimentaire dont la consommation était négligeable dans l’Union avant le 15 mai 1997. Un Novel food peut être d’origine végétale, animale, issu de la recherche, ou encore issu de la tradition de pays tiers. Il peut s’agir d’un aliment récemment développé, d’un aliment innovant ou d’un aliment produit au moyen de nouvelles technologies et de procédés inédits, ainsi que de denrées alimentaires traditionnellement consommées en dehors de l’UE.
Quelle Réglementation encadre ces ingrédients Novel Food ?
Le nouveau Règlement Novel Food (UE) n°2015/2283 est entré en vigueur depuis peu, le 1er Janvier 2018. L’entrée en application de ce règlement a donné lieu à la publication de 3 règlements d’exécution :
- Règlement (UE) 2017/2468 : définit les exigences administratives et scientifiques concernant les aliments traditionnels venant de pays tiers
- Règlement (UE) 2017/2469 : définit les exigences administratives et scientifiques concernant l’article 10 du règlement novel food
- Règlement (UE) 2017/2470 : regroupe sous la forme d’une liste les Novel Foods autorisés et leurs spécifications. Cette liste s’étoffera au fur et à mesure des autorisations


Le nouveau règlement Novel food en bref
Ce nouveau règlement a pour objectif de simplifier les procédures Novel food et de favoriser l’innovation.
Le système d’autorisation est devenu centralisé, l’évaluation scientifique des dossiers est réalisée par l’EFSA et la Commission européenne base ensuite ses décisions d’autorisation sur les avis rendus par l’EFSA. Les autorisations sont désormais génériques, ainsi tout industriel peut commercialiser un novel food autorisé s’il respecte bien les spécifications de l’ingrédient, les conditions d’utilisation et les mentions d’étiquetage. Certaines autorisations peuvent néanmoins être conférées avec protection des données pendant 5 ans pour l’opérateur ayant fourni les données scientifiques qui ont permis d’évaluer la sécurité de l’ingrédient.
Le système d’autorisation est devenu centralisé, l’évaluation scientifique des dossiers est réalisée par l’EFSA et la Commission européenne base ensuite ses décisions d’autorisation sur les avis rendus par l’EFSA. Les autorisations sont désormais génériques, ainsi tout industriel peut commercialiser un novel food autorisé s’il respecte bien les spécifications de l’ingrédient, les conditions d’utilisation et les mentions d’étiquetage. Certaines autorisations peuvent néanmoins être conférées avec protection des données pendant 5 ans pour l’opérateur ayant fourni les données scientifiques qui ont permis d’évaluer la sécurité de l’ingrédient.
Qu’en est-il des ingrédients traditionnels dans un pays tiers?
Cette procédure d’évaluation a été simplifiée. Il faut apporter la preuve que l’aliment est consommé de manière historique et sûre depuis au moins 25 ans dans le pays tiers et qu’aucune réserve quant à sa qualité n’a été émise par un Etat membre ou EFSA. L’aliment pourra alors être commercialisé sur le marché européen sur la base d’une notification réalisée par l’industriel.
Comment savoir si un ingrédient est Novel Food ?
Trois sources d’informations, présentes sur le site de la Commission européenne, peuvent être utilisées pour une première évaluation du statut Novel food ou non d’un ingrédient :
- Le Catalogue Novel Food (non exhaustif et sans valeur règlementaire) : il s’agit d’une liste d’ingrédients sur lesquels les Etats Membres et la Commission Européenne ont échangé des informations.
- Le Règlement (UE) n° 2017/2470 : Texte règlementaire qui liste toutes les autorisations Novel Food, avec leurs spécifications.
- Les demandes d’autorisation en cours : elles sont répertoriées sur le site de la Commission.
D’autres sources peuvent également être utilisées (Guide de consommation significative, Guide de substantielle équivalence, recommandation de la Commission du 29 juillet 1997, avis ANSES…). En cas de doute sur le statut réglementaire du produit, l’exploitant peut déposer une demande de consultation auprès de l’Etat membre où il a l’intention de commercialiser le produit en premier lieu. L’Etat membre va alors déterminer si l’ingrédient relève du statut Novel Food ou non et vérifier la validité de la demande.
